Feature Update: Visibility of Reads Based on SAM Flags
SAM flags are part of the Sequence Alignment Map (SAM) file format. These flags are bitwise values that describe various properties of a read in a SAM file, such as whether it is paired, mapped, reversed, or a duplicate. The BAM files, which are binary versions of the SAM files, retain these flags as well. Their values can control the visibility of the aligned reads in Persephone.
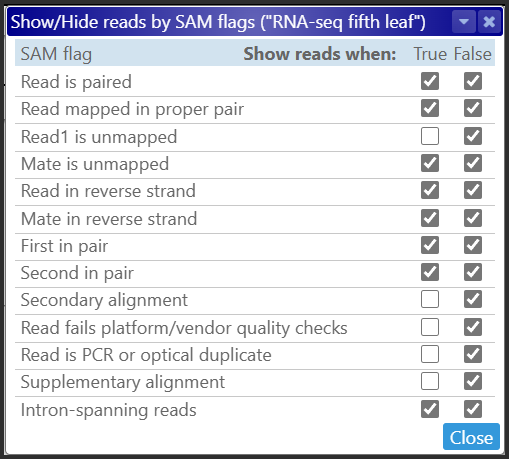
We introduced a new interface that enables users to choose which reads to display based on their flag values. This feature offers great flexibility. There may be times when you want to display perfectly aligned reads only. For this, you can show the reads where the “problem” flags are set to False. You may want to read the corresponding line with the check boxes as “Show reads where the ‘problem’ flag is false”. For instance:

At other times, you may need to focus only on those reads that have a problem, so that you can identify the source of the issue.
If both check boxes are checked, the value of the flag will have no effect on the read visibility. Apparently, the situation when both check boxes are checked off does not make much sense – no read will be shown. So, this combination of the check boxes is disabled.
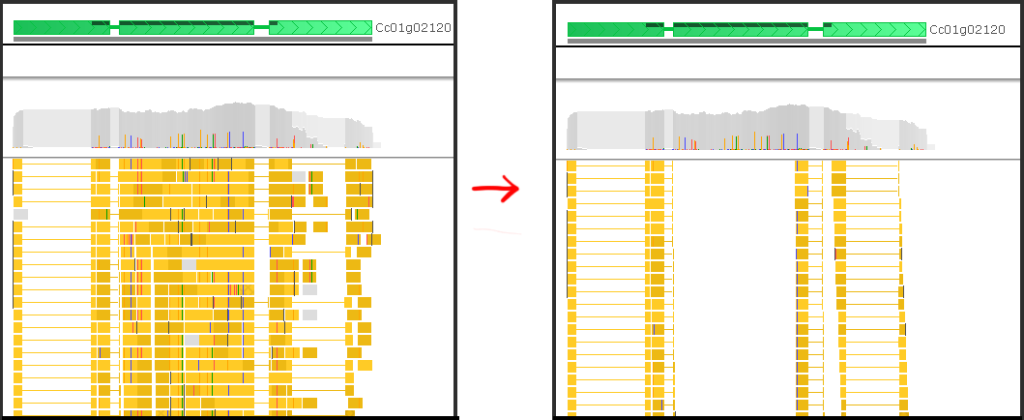
Sometimes, when studying splicing patterns, the intron-spanning reads serve as crucial evidence, while the others can obscure the picture. You can clarify the image by unchecking the False check box for Intron-spanning reads, which is equivalent to the instruction “Hide the reads where intron-spanning is set to false”.